Research
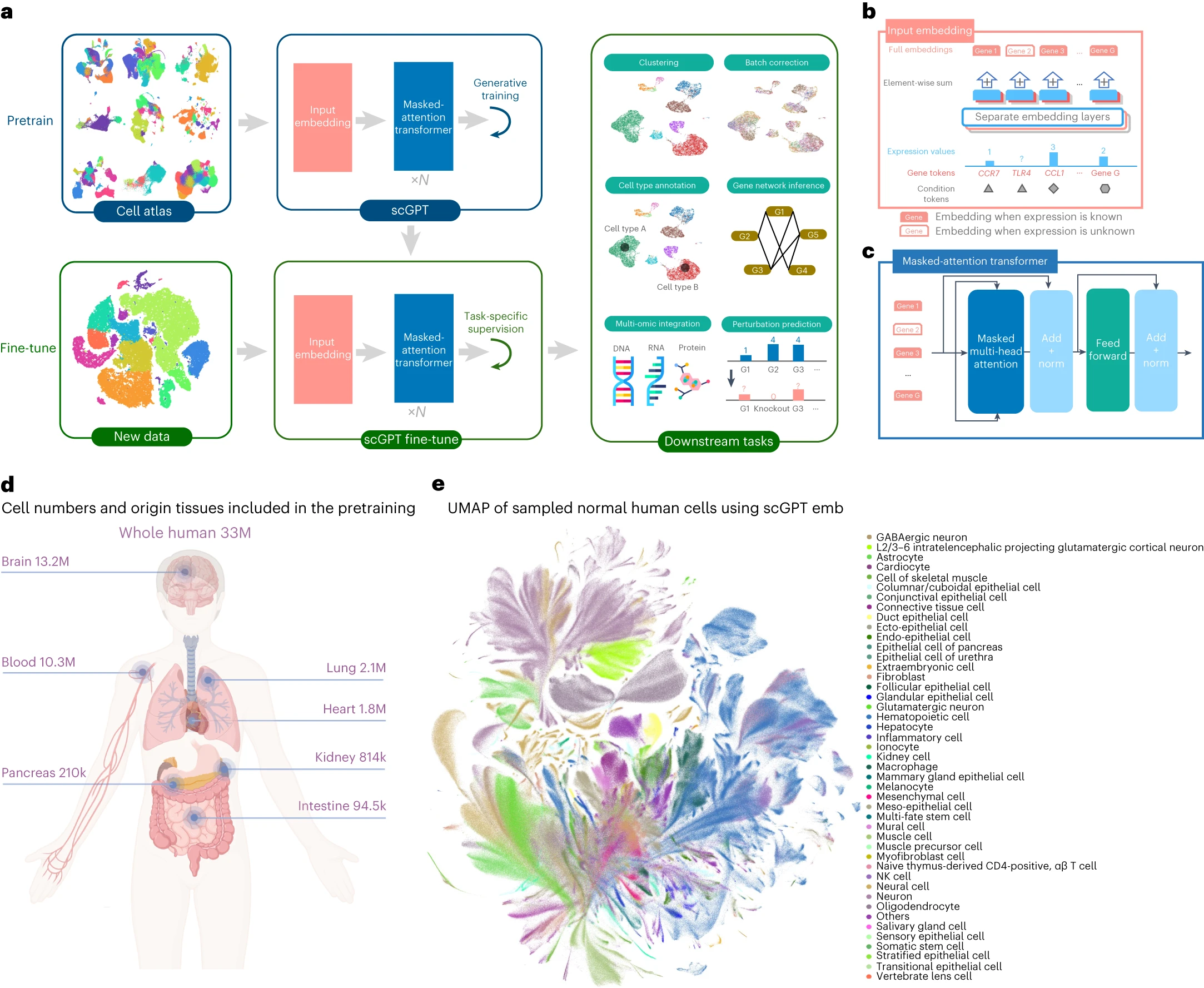
scGPT: toward building a foundation model for single-cell multi-omics using generative AI
Generative pretrained models have achieved remarkable success in various domains such as language and computer vision. Specifically, the combination of large-scale diverse datasets and pretrained transformers has emerged as a promising approach for developing foundation models. Drawing parallels between language and cellular biology (in which texts comprise words; similarly, cells are defined by genes), our study probes the applicability of foundation models to advance cellular biology and genetic research. Using burgeoning single-cell sequencing data, we have constructed a foundation model for single-cell biology, scGPT, based on a generative pretrained transformer across a repository of over 33 million cells. Our findings illustrate that scGPT effectively distills critical biological insights concerning genes and cells. Through further adaptation of transfer learning, scGPT can be optimized to achieve superior performance across diverse downstream applications. This includes tasks such as cell type annotation, multi-batch integration, multi-omic integration, perturbation response prediction and gene network inference.
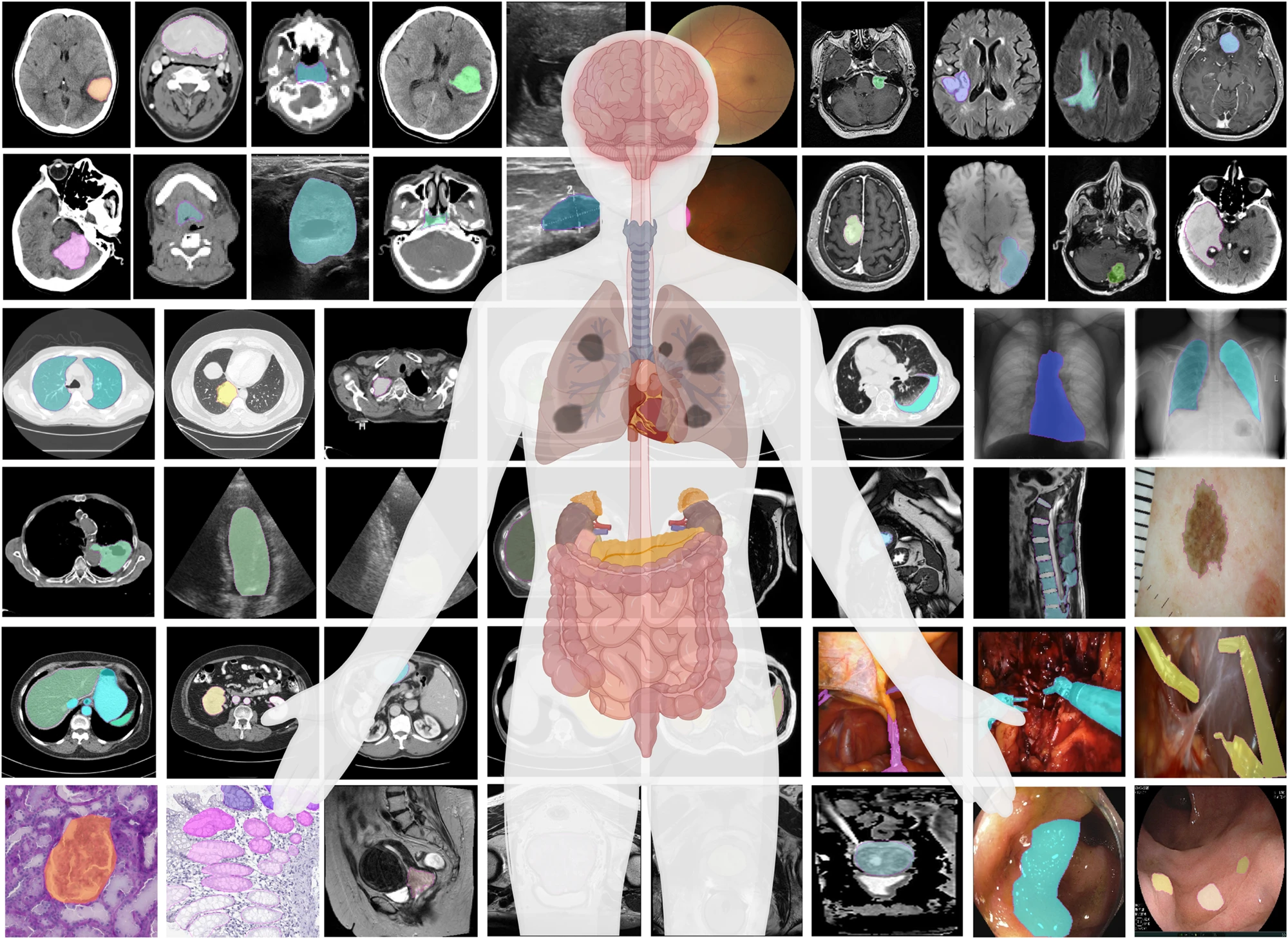
Segment anything in medical images
Medical image segmentation is a critical component in clinical practice, facilitating accurate diagnosis, treatment planning, and disease monitoring. However, existing methods, often tailored to specific modalities or disease types, lack generalizability across the diverse spectrum of medical image segmentation tasks. Here we present MedSAM, a foundation model designed for bridging this gap by enabling universal medical image segmentation. The model is developed on a large-scale medical image dataset with 1,570,263 image-mask pairs, covering 10 imaging modalities and over 30 cancer types. We conduct a comprehensive evaluation on 86 internal validation tasks and 60 external validation tasks, demonstrating better accuracy and robustness than modality-wise specialist models. By delivering accurate and efficient segmentation across a wide spectrum of tasks, MedSAM holds significant potential to expedite the evolution of diagnostic tools and the personalization of treatment plans.
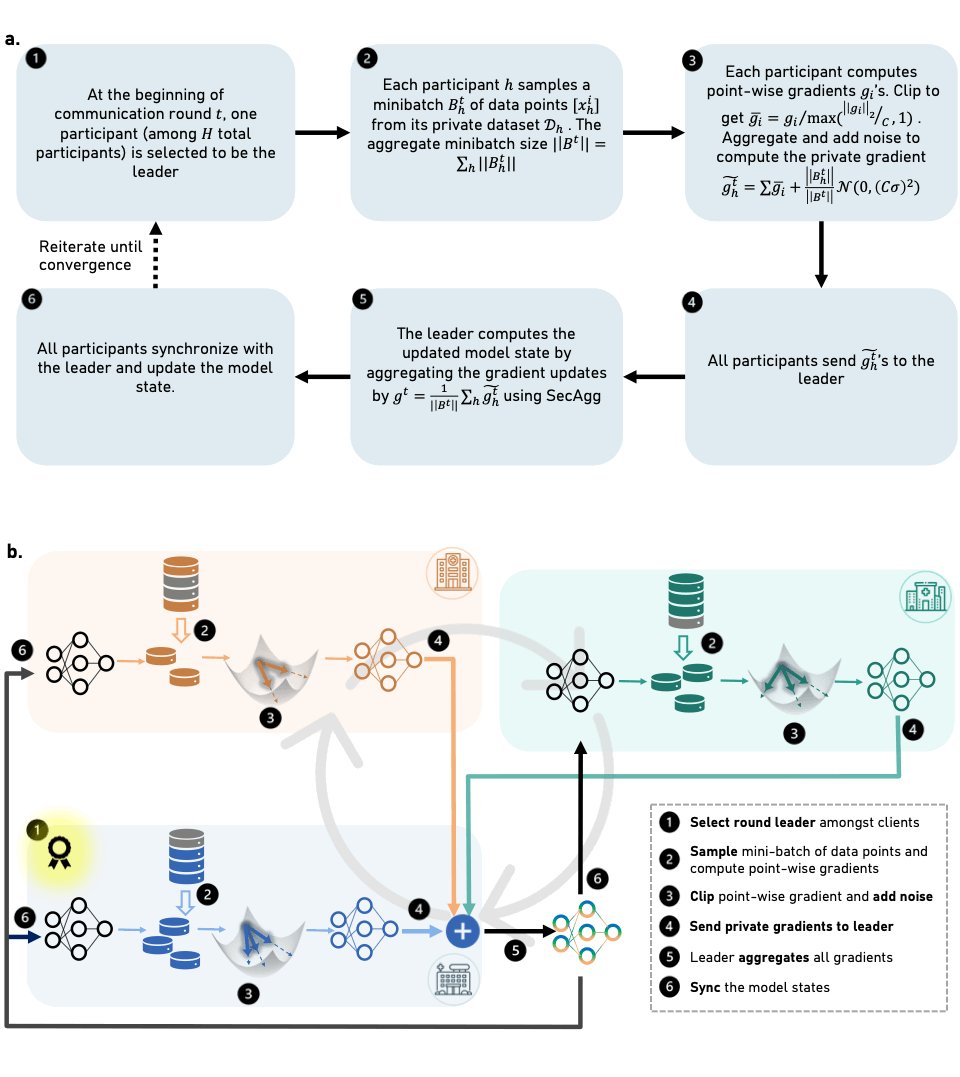
Decentralised, collaborative, and privacy-preserving machine learning for multi-hospital data
Machine Learning (ML) has demonstrated its great potential on medical data analysis. Large datasets collected from diverse sources and settings are essential for ML models in healthcare to achieve better accuracy and generalizability. Sharing data across different healthcare institutions or jurisdictions is challenging because of complex and varying privacy and regulatory requirements. Hence, it is hard but crucial to allow multiple parties to collaboratively train an ML model leveraging the private datasets available at each party without the need for direct sharing of those datasets or compromising the privacy of the datasets through collaboration.
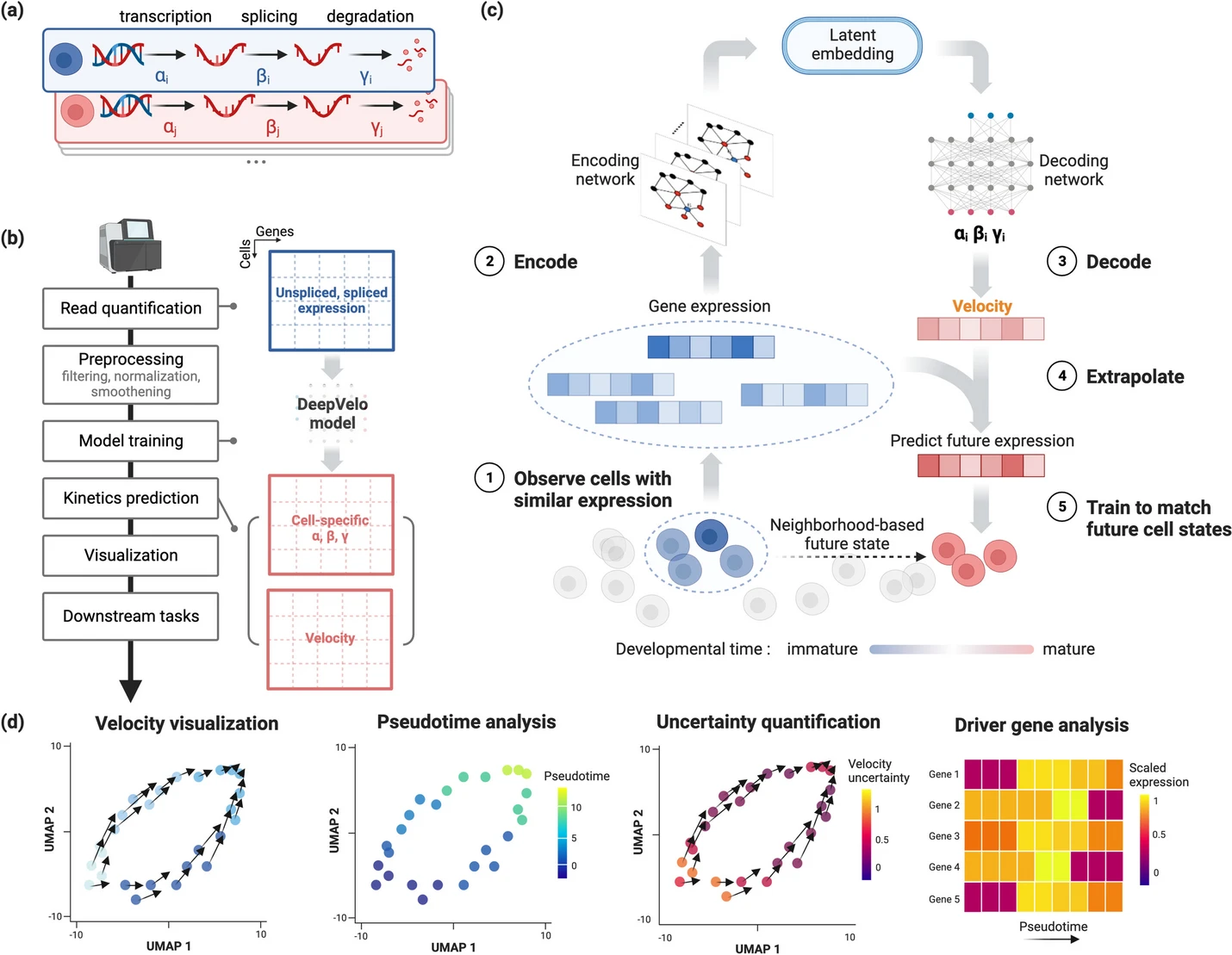
DeepVelo: deep learning extends RNA velocity to multi-lineage systems with cell-specific kinetics
Existing RNA velocity estimation methods strongly rely on predefined dynamics and cell-agnostic constant transcriptional kinetic rates, assumptions often violated in complex and heterogeneous single-cell RNA sequencing (scRNA-seq) data. Using a graph convolution network, DeepVelo overcomes these limitations by generalizing RNA velocity to cell populations containing time-dependent kinetics and multiple lineages. DeepVelo infers time-varying cellular rates of transcription, splicing, and degradation, recovers each cell’s stage in the differentiation process, and detects functionally relevant driver genes regulating these processes. Application to various developmental and pathogenic processes demonstrates DeepVelo’s capacity to study complex differentiation and lineage decision events in heterogeneous scRNA-seq data.
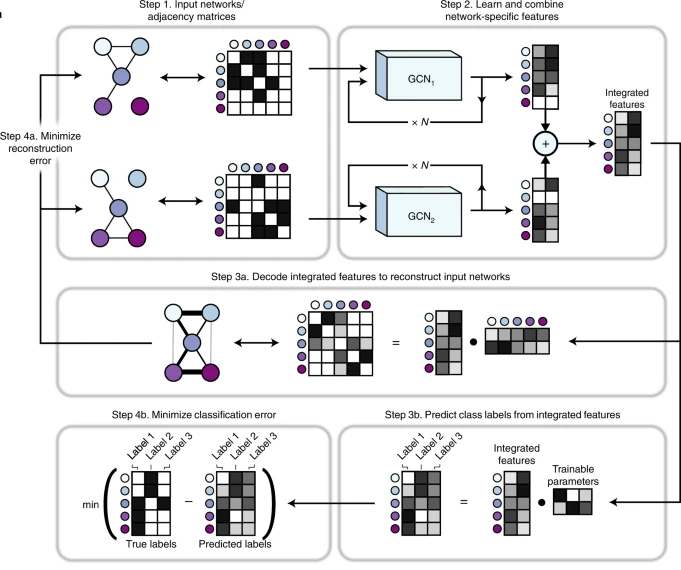
BIONIC: biological network integration using convolutions
Biological networks constructed from varied data can be used to map cellular function, but each data type has limitations. Network integration promises to address these limitations by combining and automatically weighting input information to obtain a more accurate and comprehensive representation of the underlying biology. We developed a deep learning-based network integration algorithm that incorporates a graph convolutional network framework. Our method, BIONIC (Biological Network Integration using Convolutions), learns features that contain substantially more functional information compared to existing approaches. BIONIC has unsupervised and semisupervised learning modes, making use of available gene function annotations. BIONIC is scalable in both size and quantity of the input networks, making it feasible to integrate numerous networks on the scale of the human genome. To demonstrate the use of BIONIC in identifying new biology, we predicted and experimentally validated essential gene chemical–genetic interactions from nonessential gene profiles in yeast.
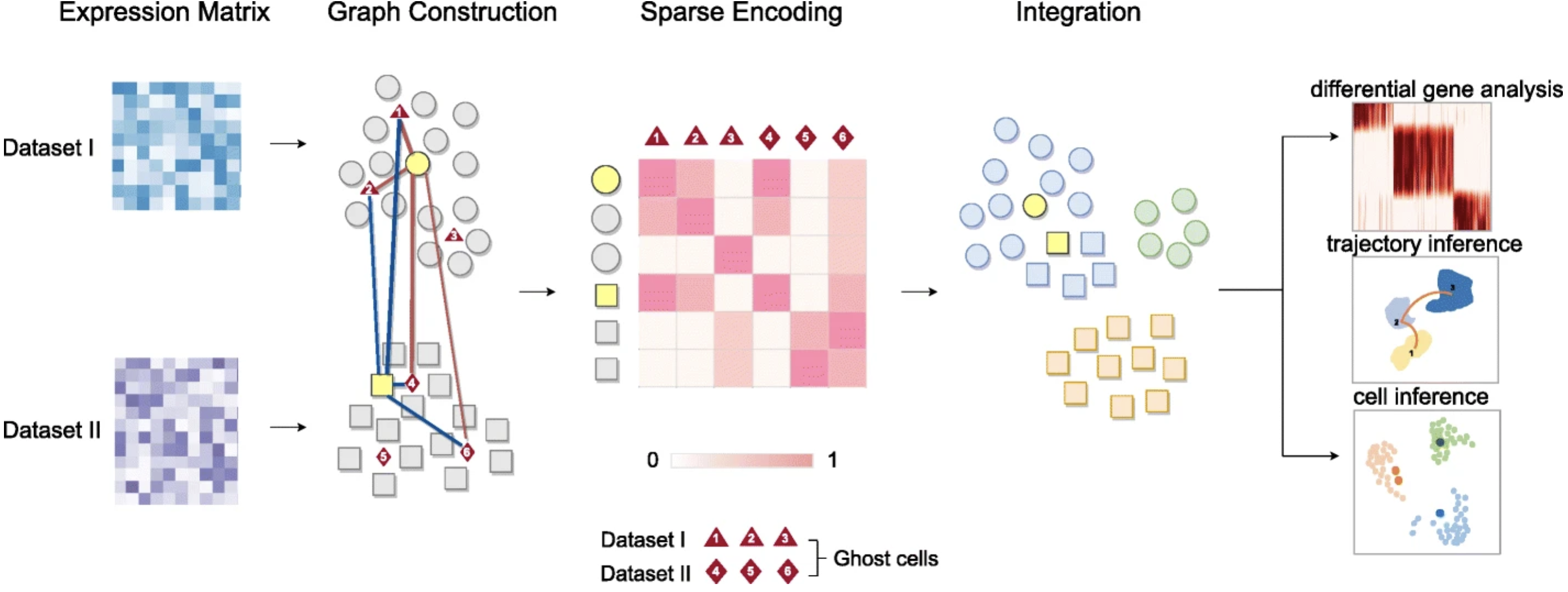
One Cell At a Time (OCAT): a unified framework to integrate and analyze single-cell RNA-seq data
Integrative analysis of large-scale single-cell RNA sequencing (scRNA-seq) datasets can aggregate complementary biological information from different datasets. However, most existing methods fail to efficiently integrate multiple large-scale scRNA-seq datasets. We propose OCAT, One Cell At a Time, a machine learning method that sparsely encodes single-cell gene expression to integrate data from multiple sources without highly variable gene selection or explicit batch effect correction. We demonstrate that OCAT efficiently integrates multiple scRNA-seq datasets and achieves the state-of-the-art performance in cell type clustering, especially in challenging scenarios of non-overlapping cell types. In addition, OCAT can efficaciously facilitate a variety of downstream analyses.
Long-term mortality risk stratification of liver transplant recipients: real-time application of deep learning algorithms on longitudinal data
Survival of liver transplant recipients beyond 1 year since transplantation is compromised by an increased risk of cancer, cardiovascular events, infection, and graft failure. Few clinical tools are available to identify patients at risk of these complications, which would flag them for screening tests and potentially life-saving interventions. In this retrospective analysis, we aimed to assess the ability of deep learning algorithms of longitudinal data from two prospective cohorts to predict complications resulting in death after liver transplantation over multiple timeframes, compared with logistic regression models.
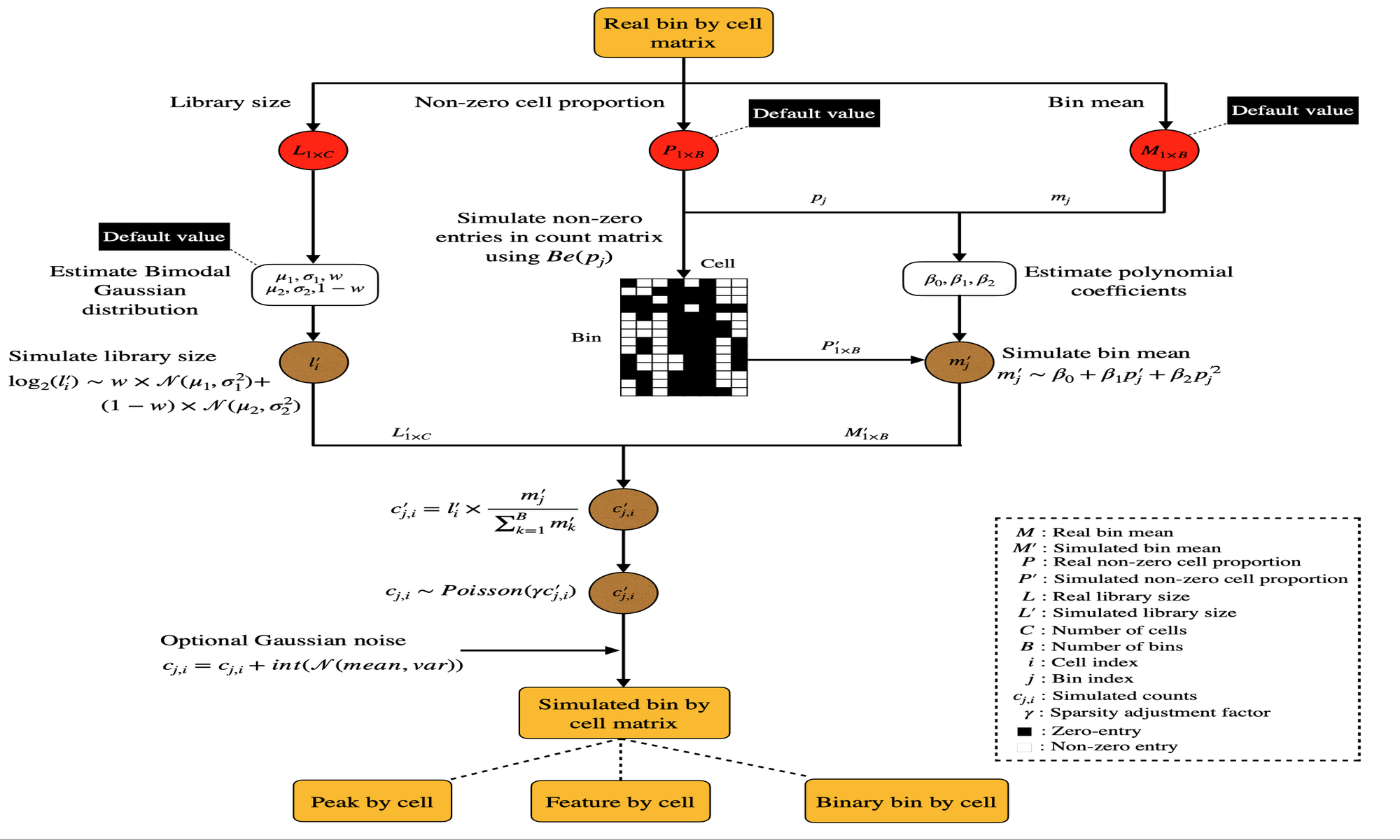
simATAC: a single-cell ATAC-seq simulation framework
Single-cell assay for transposase-accessible chromatin sequencing (scATAC-seq) identifies regulated chromatin accessibility modules at the single-cell resolution. Robust evaluation is critical to the development of scATAC-seq pipelines, which calls for reproducible datasets for benchmarking. We hereby present the simATAC framework, an R package that generates scATAC-seq count matrices that highly resemble real scATAC-seq datasets in library size, sparsity, and chromatin accessibility signals. simATAC deploys statistical models derived from analyzing 90 real scATAC-seq cell groups. simATAC provides a robust and systematic approach to generate in silico scATAC-seq samples with known cell labels for assessing analytical pipelines.
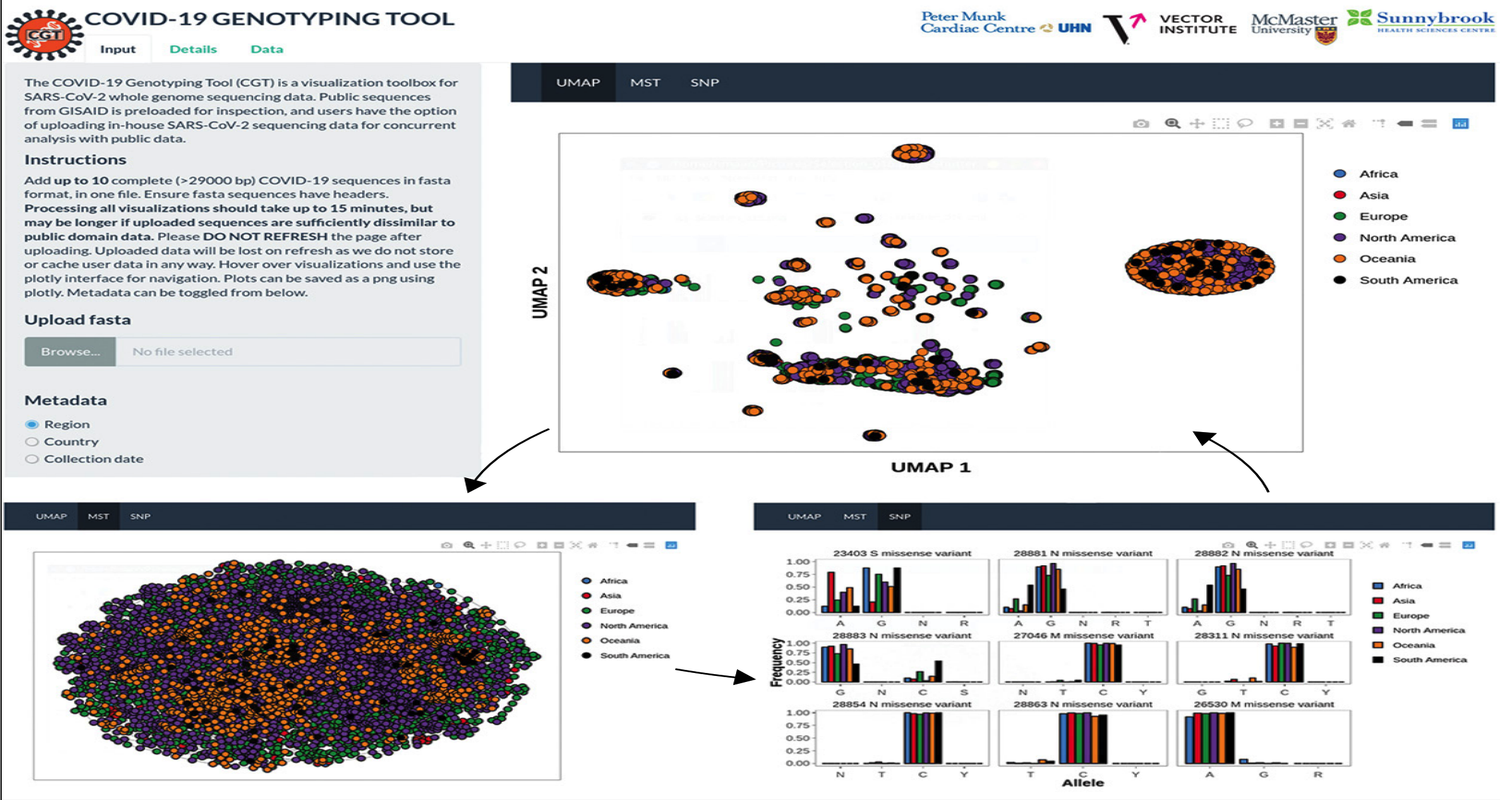
Genotyping SARS-CoV-2 through an interactive web application
The ongoing COVID-19 pandemic is the greatest health-care challenge of this generation. Early viral genome sequencing studies of small cohorts have indicated the possibility of distinct severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) genotypes. If these subtypes result in an altered virus tropism or pathogenesis in infected hosts, this could have immediate implications for vaccine design, drug development, and efforts to control the pandemic. Therefore, the genomic surveillance and characterisation of circulating viral strains is a high priority for research and development. To facilitate the epidemiological tracking of SARS-CoV-2, researchers worldwide have created various web-portals and tools, such as the Johns Hopkins University COVID-19 dashboard. An unprecedented effort to make COVID-19-related data accessible in near real-time has resulted in more than 25 000 publicly available genome sequences of SARS-CoV-2 on Global Initiative on Sharing All Influenza Data (GISAID). Although platforms to survey epidemiological data are prevalent, tools that summarise publicly available viral genome data are scarce and those that are available do not offer users the ability to analyse in-house sequencing data. To address this gap, we have developed an accessible application, the COVID-19 Genotyping Tool (CGT).
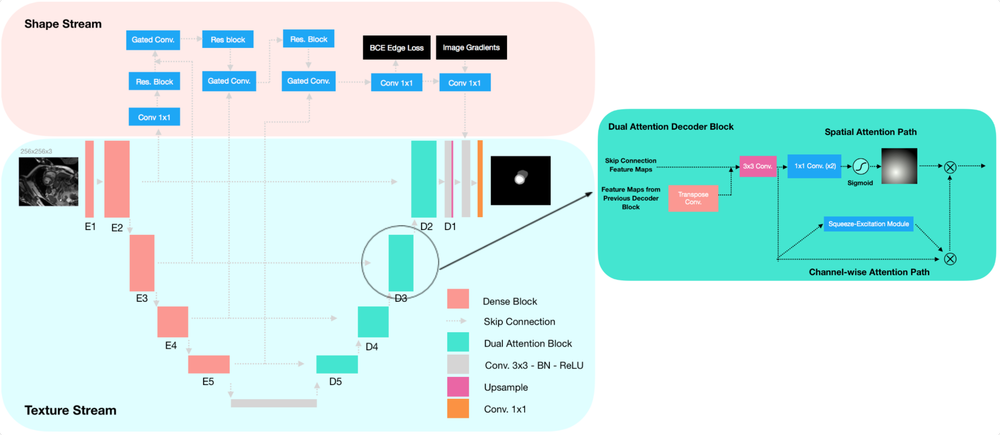
SAUNet: Shape Attentive U-Net for Interpretable Medical Image Segmentation
Medical image segmentation is a difficult but important task for many clinical operations such as cardiac bi-ventricular volume estimation. More recently, there has been a shift to utilizing deep learning and fully convolutional neural networks (CNNs) to perform image segmentation that has yielded state-of-the-art results in many public benchmark datasets. Despite the progress of deep learning in medical image segmentation, standard CNNs are still not fully adopted in clinical settings as they lack robustness and interpretability. Shapes are generally more meaningful features than solely textures of images, which are features regular CNNs learn, causing a lack of robustness. Likewise, previous works surrounding model interpretability have been focused on post hoc gradient-based saliency methods. However, gradient-based saliency methods typically require additional computations post hoc and have been shown to be unreliable for interpretability. Thus, we present a new architecture called Shape Attentive U-Net (SAUNet) which focuses on model interpretability and robustness. The proposed architecture attempts to address these limitations by the use of a secondary shape stream that captures rich shape-dependent information in parallel with the regular texture stream. Furthermore, we suggest multi-resolution saliency maps can be learned using our dual-attention decoder module which allows for multi-level interpretability and mitigates the need for additional computations post hoc.
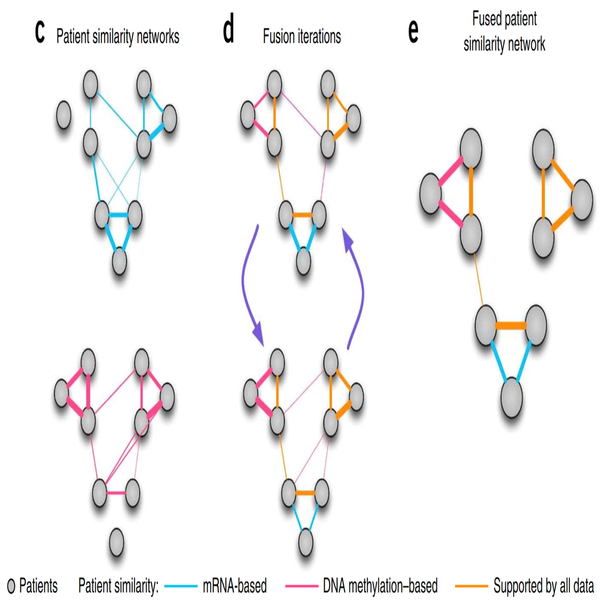
Similarity network fusion for aggregating data types on a genomic scale
Recent technologies have made it cost-effective to collect diverse types of genome-wide data. Computational methods are needed to combine these data to create a comprehensive view of a given disease or a biological process. Similarity network fusion (SNF) solves this problem by constructing networks of samples (e.g., patients) for each available data type and then efficiently fusing these into one network that represents the full spectrum of underlying data. For example, to create a comprehensive view of a disease given a cohort of patients, SNF computes and fuses patient similarity networks obtained from each of their data types separately, taking advantage of the complementarity in the data. We used SNF to combine mRNA expression, DNA methylation and microRNA (miRNA) expression data for five cancer data sets. SNF substantially outperforms single data type analysis and established integrative approaches when identifying cancer subtypes and is effective for predicting survival.
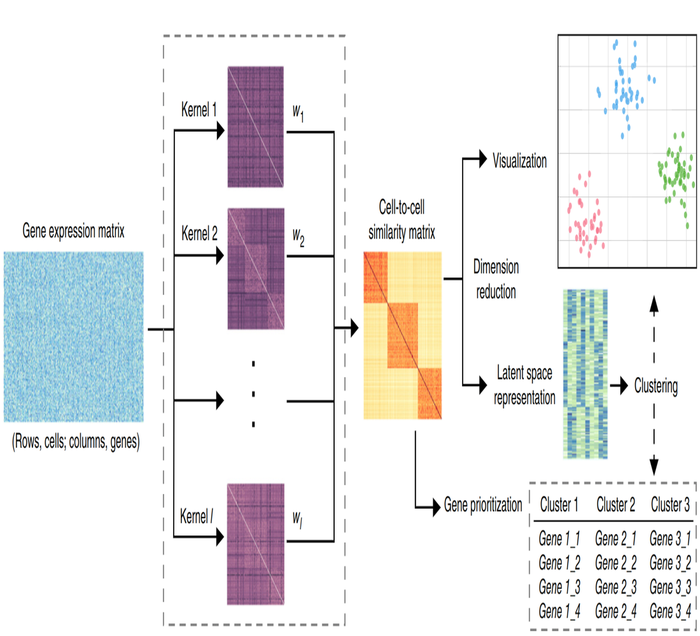
Visualization and analysis of single-cell RNA-seq data by kernel-based similarity learning
We present single-cell interpretation via multikernel learning (SIMLR), an analytic framework and software which learns a similarity measure from single-cell RNA-seq data in order to perform dimension reduction, clustering and visualization. On seven published data sets, we benchmark SIMLR against state-of-the-art methods. We show that SIMLR is scalable and greatly enhances clustering performance while improving the visualization and interpretability of single-cell sequencing data.
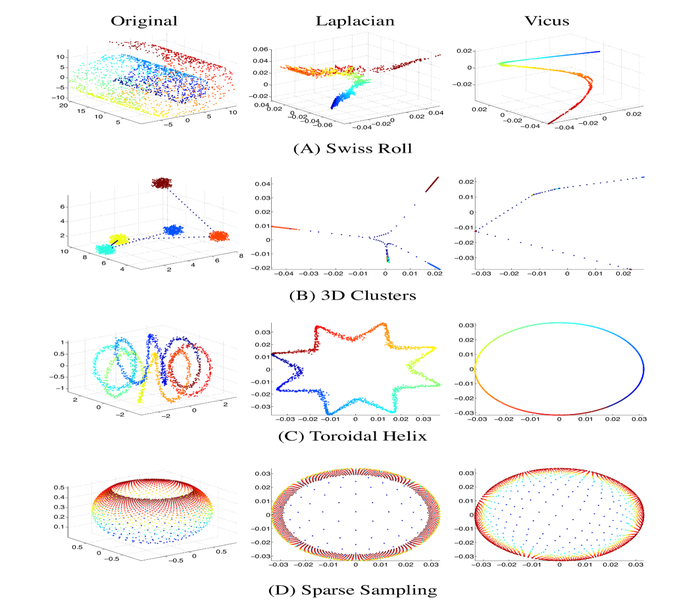
Vicus: Exploiting local structures to improve network-based analysis of biological data
Biological networks entail important topological features and patterns critical to understanding interactions within complicated biological systems. Despite a great progress in understanding their structure, much more can be done to improve our inference and network analysis. Spectral methods play a key role in many network-based applications. Fundamental to spectral methods is the Laplacian, a matrix that captures the global structure of the network. Unfortunately, the Laplacian does not take into account intricacies of the network’s local structure and is sensitive to noise in the network. These two properties are fundamental to biological networks and cannot be ignored. We propose an alternative matrix Vicus. The Vicus matrix captures the local neighborhood structure of the network and thus is more effective at modeling biological interactions. We demonstrate the advantages of Vicus in the context of spectral methods by extensive empirical benchmarking on tasks such as single cell dimensionality reduction, protein module discovery and ranking genes for cancer subtyping. Our experiments show that using Vicus, spectral methods result in more accurate and robust performance in all of these tasks.
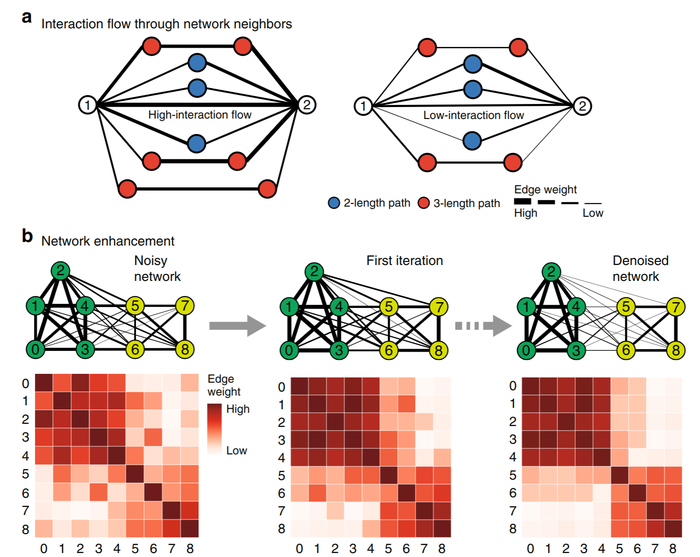
Network Enhancement: a general method to denoise weighted biological networks
Networks are ubiquitous in biology where they encode connectivity patterns at all scales of organization, from molecular to the biome. However, biological networks are noisy due to the limitations of measurement technology and inherent natural variation, which can hamper discovery of network patterns and dynamics. We propose Network Enhancement (NE), a method for improving the signal-to-noise ratio of undirected, weighted networks. NE uses a doubly stochastic matrix operator that induces sparsity and provides a closed-form solution that increases spectral eigengap of the input network. As a result, NE removes weak edges, enhances real connections, and leads to better downstream performance. Experiments show that NE improves gene–function prediction by denoising tissue-specific interaction networks, alleviates interpretation of noisy Hi-C contact maps from the human genome, and boosts fine-grained identification accuracy of species. Our results indicate that NE is widely applicable for denoising biological networks.